再発またはステージIVの非小細胞肺がんに対するアベルマブの治験
治験の募集状況は、「jRCT 臨床研究等提出・公開システム![]() 」ページでご確認ください。
」ページでご確認ください。
治験名
非小細胞肺癌を対象とした一次治療におけるアベルマブの検討
再発性またはステージIVのPD-L1陽性非小細胞肺癌の一次治療としてアベルマブと白金製剤を含む2剤併用化学療法とを比較する第3相非盲検多施設共同試験
治験概要:
再発またはステージIVのPD-L1陽性の非小細胞肺がんの一次治療を対象とした治験です。アベルマブと白金製剤を含む2剤を併用した化学療法とを比較する第3相試験です。
登録予定数は1095人。
試験デザインは、非盲検多施設共同試験。
フェーズは、第3相臨床試験。
比較する対象は
対象群1:アベルマブ
対象群2:白金製剤を含む2剤併用化学療法
で主要評価項目は、PD-L1 強陽性の患者を対象とした無増悪生存期間、全生存期間、副次的な評価項目は、PD-L1 中等度陽性または強陽性の患者を対象とした無増悪生存期間、全生存期間ほかで評価します。
疾患解説:肺がん
疾患の詳細は、「肺がんを知る」を参照ください。
治験薬:アベルマブ
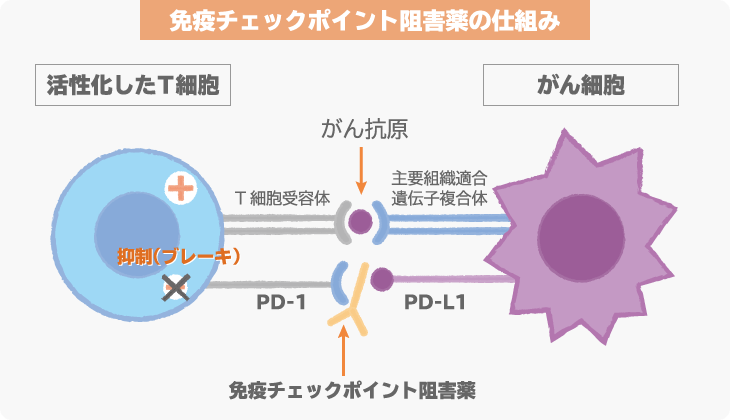
アバルマブは、抗PD-L1抗体という免疫チェックポイント阻害薬の1つです。
免疫チェックポイント阻害薬は、がんに対して、免疫細胞が本来の力を発揮できるようにする薬です。最終的には、免疫の力でがんを攻撃し、治療効果を発揮します。
がん細胞の表面に発現しているPD-L1とがん細胞を攻撃する免疫細胞(T細胞)に発現しているPD-1が結合すると、免疫細胞は、がん細胞を攻撃しなくなってしまいます。この仕組みを「免疫チェックポイント機構」といい、この仕組みが働かないように開発されたのが、免疫チェックポイント阻害薬です。
治験情報に関する注意点
治験は、治療を兼ねた臨床試験のことです。薬の元となる物質を動物実験などで有効性や安全性を確認した上で、ヒトに対して使用しても同様に安全で治療効果が予測されるもので行われますが、治験の時点ではまだ有効性や安全性が十分に確認できているわけではありません。有効性や安全性が科学的に証明された治療が、標準治療で、新しい治療が必ずしも最良の治療ではないということを理解してください。その一方で標準治療が確立していない、または薬の耐性ができ、効果が期待できる薬がなくなった患者さんにとって治験は新しい治療選択となる可能性もあります。
治験は「ヘルシンキ宣言」に基づく倫理的原則と、「医薬品の臨床試験の実施に関する基準(GCP)」を遵守して行われています。これにより、治験に参加される方の利益が損なわれることがないよう、安全な手続きで治験は進められます。
治験情報を探すとき、治験を受けたいと思ったときは、まず治験とはどのようなものなのかを理解してください。
がんの治験情報をお探しの方に知ってほしい5つのこと
※ここに掲載した情報は、jRCT 臨床研究等提出・公開システム に登録された情報を元にし、がんプラスが独自に記事としてまとめ、提供しています。
※QLife「がん治験情報サービス」でご案内している治験とは異なります。
試験概要詳細
| 試験の名称 | 再発性またはステージIVのPD-L1 陽性非小細胞肺癌の一次治療としてアベルマブ(MSB0010718C)と白金製剤を含む2剤併用化学療法とを比較する第III相非盲検多施設共同試験 |
| 試験の概要 | 再発性またはステージIVのPD-L1 陽性非小細胞肺癌の一次治療としてアベルマブ(MSB0010718C)と白金製剤を含む2剤併用化学療法とを比較する第III相非盲検多施設共同試験 |
| 疾患名 | 非小細胞肺癌の一次治療 |
| 試験薬剤名 | アベルマブ |
| 用法・用量 | アベルマブ:10 mg/kg の用量で1 時間の点滴静注により1 週間に1 回、12 週間連続投与した後、同量 を2週間に1 回投与する |
| 試験薬剤名 | ペメトレキセド(500mg/m2)とシスプラチン(75 mg/m2)またはカルボプラチン(6mg/mL*min) |
| 用法・用量 | 点滴静注により3週間に1回投与する |
| 試験のフェーズ | フェーズ3(第3相臨床試験) |
| 試験のデザイン | |
| 目標症例数 | 1095 |
| 適格基準 |
|
| 除外基準 |
|
| 主要な評価項目 | RECIST1.1に基づく独立レビュー委員会(IRC)評価によるPD-L1 強陽性のNSCLC患者を対象に、無増悪生存期間(PFS)について、白金製剤を含む2剤併用療法に対するアベルマブの優越性を示すことである。 |
| 主要な評価方法 | [期間:無作為割付け日からPDまたは死亡日で39ヶ月目まで評価される。][指定された安全性問題:No.] PFSは、無作為割付け日から最初のPD(IRCが判定)、またはPDの記録がない場合は死因を問わない死亡日のいずれか早い方までの期間と定義する。 |
| 主要な評価項目 | RECIST1.1に基づく独立レビュー委員会(IRC)評価によるPD-L1強陽性のNSCLC患者を対象に、全生存期間(OS)について、白金製剤を含む2剤併用療法に対するアベルマブの優越性を示すことである |
| 主要な評価方法 | [期間:無作為割付け日からPDまたは死亡日で49ヶ月目まで評価される。][指定された安全性問題:No.] OSは、無作為割付け日から死因を問わない死亡日までの期間と定義する。 |
| 副次的な評価項目 | RECIST1.1に基づくIRC評価によるPD-L1中等度陽性または強陽性のNSCLC患者を対象に、PFSについて、アベルマブの優越性を示す。 |
| 副次的な評価方法 | [期間:無作為割付け日からPDまたは死亡日で39ヶ月目まで評価される。][指定された安全性問題:No.] PFSは、無作為割付け日から最初のPD(IRCが判定)、またはPDの記録がない場合は死因を問わない死亡日のいずれか早い方までの期間と定義する。 |
| 副次的な評価項目 | RECIST1.1に基づくIRC評価によるPD-L1中等度陽性または強陽性のNSCLC患者を対象に、OSについて、アベルマブの優越性を示す。 |
| 副次的な評価方法 | [期間:無作為割付け日からPDまたは死亡日で49ヶ月目まで評価される。][指定された安全性問題:No.] OSは、無作為割付け日から死因を問わない死亡日までの期間と定義する。 |
| 副次的な評価項目 | IRC判定による最良総合効果(BOR) |
| 副次的な評価方法 | [期間:無作為割付け日から39ヶ月目まで][指定された安全性問題:No.] BORはRECIST1.1に基づいて決定される。CR:標的病変及び非標的病変が全て消失すること。PR:全病変のベースライン長径和30%以上減少すること。安定(SD):PDとするには腫瘍の増大が不十分で、PRとするには腫瘍の縮小が不十分であること。 |
| 副次的な評価項目 | RECIST1.1に基づく奏効期間(DOR) |
| 副次的な評価方法 | [期間:無作為割付け日から39ヶ月目まで][指定された安全性問題:No.] RECIST1.1に基づくDORは、奏効が確定した各被験者について、CRまたはPRが認められた最初の評価日から、PDが最初に認められた日、または最終腫瘍評価後12週間以内の死亡日までのいずれか早い方までの期間と定義する。CR:標的病変及び非標的病変が全て消失すること。PR:全病変のベースライン長径和が30%以上減少すること。安定(SD):PDとするには腫瘍の増大が不十分で、PRとするには腫瘍の縮小が不十分であること。PDはベースライン時に記録された最小の長径和と比較して標的病変の最長径の和が20%以上増加、あるいは1つ以上の新病変が出現すること。 |
| 副次的な評価項目 | 被験者報告アウトカム:European Quality of Life 5-dimensions 5-levelquestionnaire(EQ-5D-5L)健康関連質問票からの変化 |
| 副次的な評価方法 | [期間:ベースライン時から39ヶ月目まで][指定された安全性問題:No.] EQ-5D-5L健康関連質問票は簡単な記述プロフィールと単一指標値によって健康状態を測る。EQ-5D-5Lは移動の程度、身の回りの管理、ふだんの活動、痛み/不快感、不安/ふさぎ込みの観点から健康を定義し、全般的健康プロフィールに組み入れられる。これらのプロフィールは1対1のマッチングを用いて連続する単一スコアに換算される。最も低いスコアは-0.59(歩き回れない、自分で身体を洗ったり着替えをすることができない、ふだんの活動を行うことができない、極度の痛みや不快感がある、極度に不安あるいはふさぎ込んでいる)で、一番高いスコアは1.00である(5つの全ての項目において何の問題もない)。 |
| 副次的な評価項目 | 欧州がん研究・治療機構の生活の質に関する質問票(EORTC QLQ-C30)の全般的健康のベースライン時からの変化 |
| 副次的な評価方法 | [期間:ベースライン時から49ヶ月目まで][指定された安全性問題:No.] EORTC QLQ-C30は癌患者の全般的な生活の質(QoL)を評価するための30項目から成る質問票である。全般的健康(GHS)、5つの活動性尺度(身体的活動性、役割活動性、認識する活動性、精神的活動性、社会的活動性)、及び9の身体症状尺度/項目(疲れ、悪心・嘔吐、痛み、息切れ、不眠、食欲不振、便秘、下痢、経済状態)の15領域から成る。EORTC QLQ-C30 GHS/QoLスコアは0から100で表される。スコアが高いと、GHS/QoLがより良い状態であるを示し、スコアが0であれば健康状態及びQoLが非常に良くないことを示す。スコアが100であれば全般的健康状態及びQoLが優れていることを示す。 |
| 副次的な評価項目 | 欧州がん研究・治療機構の生活の質に関する質問票(EORTC QLQ-LC13)のベースライン時からの変化 |
| 副次的な評価方法 | [期間:ベースライン時から49ヶ月目まで][指定された安全性問題:No.]. EORTC QLQ-LC13は肺癌特有の症状や化学療法及び放射線療法の特徴的な副作用に関する13の質問項目から成る。呼吸困難に関する1つの複数評価尺度と10つの単一症状と副作用(咳、喀血、伝染性膿瘡、呼吸困難、神経障害、脱毛、痛み止めの薬)がある。スコアの範囲:0(症状領域または単一項目での負担が全くない)から100(症状領域及び単一項目での負担が大きい)で表される。 |
| 副次的な評価項目 | 項目での負担が全くない)から100(症状領域及び単一項目での負担が大きい)で表される。 ・米国がん研究所有害事象共通用語規準(NCI-CTCAE)v4.03に基づく治療下で発現したAE(TEAE)を発現した被験者数。 |
| 副次的な評価方法 | [期間:治験薬の初回投与から最終投与後30日目まで][指定された安全性問題:Yes] TEAEは治験薬の初回投与と最終投与後30日目までに発現した有害事象で、投与前には見られなかったもの、もしくは投与前の状態に比べて悪化した事象と定義する。 |
| 副次的な評価項目 | NCI-CTCAE v4.03に従って分類された安全性に関する臨床検査に異常がある被験者の数。 |
| 副次的な評価方法 | [期間:治験薬初回投与から最終投与後30日目まで][指定された安全性問題:No.] |
| 副次的な評価項目 | バイタルサイン、身体的診察及びECOGPSがある被験者数。 |
| 副次的な評価方法 | [期間:治験薬の初回投与から最終投与後30日目までの治療][指定された安全性問題:Yes.] |
| 副次的な評価項目 | 12誘導心電図の異常がある被験者数。 |
| 副次的な評価方法 | [期間:スクリーニング及び治験終了visit][指定された安全性問題:Yes] |
| 予定試験期間 | ~2024年9月 |
出典:臨床研究等提出・公開システムより