高リスク皮膚有棘細胞がんに対するセミプリマブの治験
治験の募集状況は、「jRCT 臨床研究等提出・公開システム![]() 」ページでご確認ください。
」ページでご確認ください。
治験名
高リスク皮膚有棘細胞がん患者を対象に、手術および放射線療法後のセミプリマブによる補助療法とプラセボとを比較する無作為化、プラセボ対照、二重盲検試験
治験概要:
高リスク皮膚有棘細胞がんに対する治験。手術および放射線治療を受けた患者さんが対象です。
セミプリマブとプラセボを比較して、有効性と安全性で評価する臨床試験です。
登録予定数は、412人。
フェーズは、第3相臨床試験。
試験デザインは、ランダム化、プラセボ対照、二重盲検。
試験群:セミプリマブ
対照群:プラセボ
無病生存率、全生存期間、有害事象発現率、有害事象重症度、死亡率などで評価します。
疾患解説:有棘細胞がん
有棘細胞がんは、日本人に多くみられる皮膚がんの1つで、表皮の中間層にある有棘層の細胞ががん化した悪性腫瘍です。
有棘細胞がんの原因として、最も考えられるのが紫外線です。短期間に浴びる大量の紫外線はもちろん、長年の蓄積の影響でも発生する可能性があります。また、子宮頸がんの発症原因として知られるヒトパピローマウイルス(HPV)も原因の1つとされています。そのほか、やけどや外傷、おでき、皮膚潰瘍、長期の褥瘡、放射線治療後の慢性放射線皮膚炎なども原因となります。主な症状は、発生部位や原因によって異なります。
有棘細胞がんの特徴は、大きくて不揃いな紅色をしており、盛り上がったかたまりが崩れたような形をしています。大きくなるとカリフラワーのように見えます。
治験薬:セミプリマブ
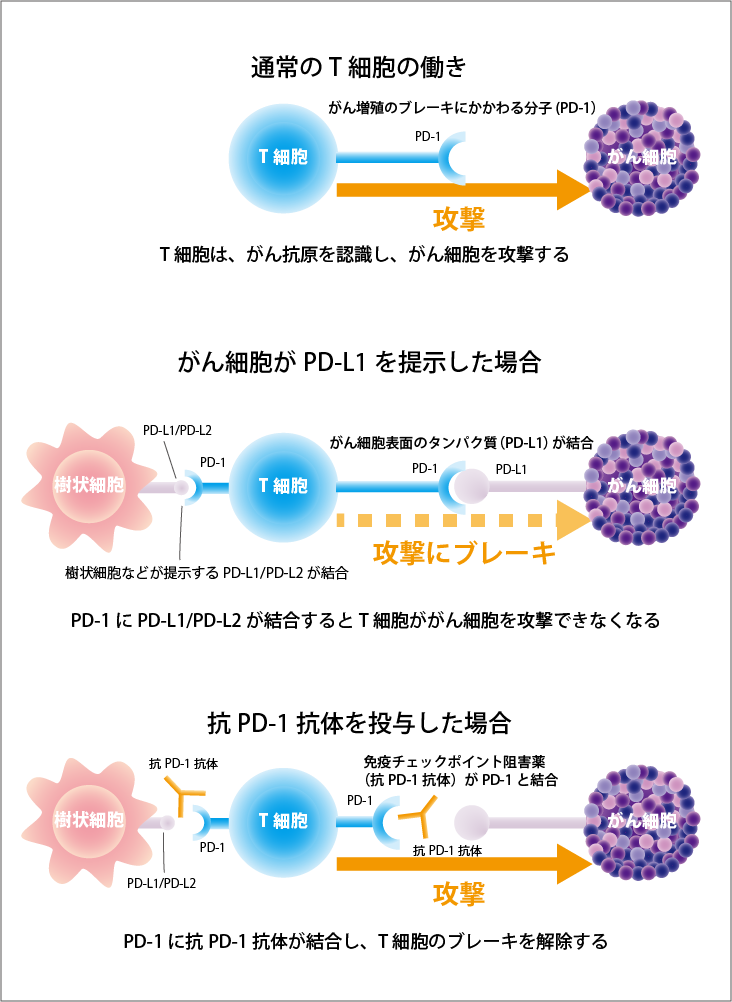
セミプリマブは、抗PD-1抗体という免疫チェックポイント阻害薬の1つです。
免疫チェックポイント阻害薬は、がんに対して、免疫細胞が本来の力を発揮できるようにする薬です。最終的には、免疫の力でがんを攻撃し、治療効果を発揮します。
がん細胞の表面に発現しているPD-L1とがん細胞を攻撃する免疫細胞(T細胞)に発現しているPD-1が結合すると、免疫細胞は、がん細胞を攻撃しなくなってしまいます。この仕組みを「免疫チェックポイント機構」といい、この仕組みが働かないように開発されたのが、免疫チェックポイント阻害薬です。
治験情報に関する注意点
治験は、治療を兼ねた臨床試験のことです。薬の元となる物質を動物実験などで有効性や安全性を確認した上で、ヒトに対して使用しても同様に安全で治療効果が予測されるもので行われますが、治験の時点ではまだ有効性や安全性が十分に確認できているわけではありません。有効性や安全性が科学的に証明された治療が、標準治療で、新しい治療が必ずしも最良の治療ではないということを理解してください。その一方で標準治療が確立していない、または薬の耐性ができ、効果が期待できる薬がなくなった患者さんにとって治験は新しい治療選択となる可能性もあります。
治験は「ヘルシンキ宣言」に基づく倫理的原則と、「医薬品の臨床試験の実施に関する基準(GCP)」を遵守して行われています。これにより、治験に参加される方の利益が損なわれることがないよう、安全な手続きで治験は進められます。
治験情報を探すとき、治験を受けたいと思ったときは、まず治験とはどのようなものなのかを理解してください。
がんの治験情報をお探しの方に知ってほしい5つのこと
※ここに掲載した情報は、jRCT 臨床研究等提出・公開システム に登録された情報を元にし、がんプラスが独自に記事としてまとめ、提供しています。
※QLife「がん治験情報サービス」でご案内している治験とは異なります。
試験概要詳細
| 試験の名称 | 高リスク皮膚有棘細胞癌患者を対象に、手術及び放射線療法後のCEMIPLIMABによる補助療法とプラセボとを比較する無作為化、プラセボ対照、二重盲検試験 |
| 試験の概要 | 本治験の主要目的は、手術及び放射線療法(RT)後にcemiplimabによる補助療法又はプラセボ投与を受けた高リスク皮膚有棘細胞癌(CSCC)の被験者の無病生存期間(DFS)を比較することである 本治験の副次目的は以下の通りである 手術及びRT後にcemiplimabによる補助療法又はプラセボ投与を受けた高リスクCSCCの被験者の全生存期間(OS)を比較すること 被験者の手術及びRT後の局所領域再発までの期間(FFLRR)に対するcemiplimabによる補助療法とプラセボの効果を比較すること 被験者の手術及びRT後の遠隔再発までの期間(FFDR)に対するcemiplimabによる補助療法とプラセボの効果を比較すること 手術及びRT後の二次性の原発性皮膚有棘細胞癌(SPT)の累積発現率に対するcemiplimabによる補助療法とプラセボの効果を比較すること 手術及びRT後の高リスクCSCCの被験者におけるcemiplimabによる補助療法とプラセボの安全性を評価すること |
| 疾患名 | 皮膚有棘細胞癌 |
| 試験薬剤名 | Cemiplimab |
| 用法・用量 | 30分かけて静脈内(IV)投与する |
| 対照薬剤名 | プラセボ |
| 用法・用量 | 30分かけてIV投与する |
| 試験のフェーズ | フェーズ3/phase3 |
| 試験のデザイン | ランダム化 プラセボ対照 二重盲検 |
| 目標症例数 | 412 |
| 適格基準 |
|
| 除外基準 |
|
| 主要な評価項目 | 有効性/efficacy |
| 主要な評価方法 | DFSは無作為化の時点から疾患の再発(局所、領域及び/又は遠隔)の最初の記録又は死因を問わない死亡までの期間と定義する。【評価期間:最長54ヵ月】 腫瘍の再発又は死亡に至っていない患者のDFSの打ち切り日は、疾患の最終評価日とする |
| 副次的な評価項目 | 有効性/efficacy |
| 副次的な評価方法 | OSは無作為化の時点から死亡日までと定義する。死亡していない被験者の打ち切り日は、生存が判明している最後の日とする。【評価期間:最長78ヵ月】 |
| 副次的な評価項目 | 有効性/efficacy |
| 副次的な評価方法 | FFLRRは無作為化の時点から局所領域再発(LRR)が最初に認められた日までと定義する。LRRが認められないまま死亡した被験者のデータ打ち切り日は、死亡日とする。【評価期間:最長54ヵ月】 LRRが認められず死亡していない被験者のFFLRRの打ち切り日は、疾患の最終評価日とする |
| 副次的な評価項目 | 有効性/efficacy |
| 副次的な評価方法 | FFDRは無作為化の時点から遠隔再発(DR)が最初に認められた日までと定義する。DRが認められないまま死亡した被験者のデータ打ち切り日は、死亡日とする。【評価期間:最長54ヵ月】 DRが認められず死亡していない被験者のFFDRの打ち切り日は、疾患の最終評価日とする |
| 副次的な評価項目 | 有効性/efficacy |
| 副次的な評価方法 | 各被験者における、無作為化から主要評価項目の最初の事象の発生又は治験終了までのSPTの累積発現件数 【評価期間:最長54ヵ月】 |
| 副次的な評価項目 | 安全性/safety |
| 副次的な評価方法 | 治験薬投与下で発現した有害事象(TEAE)の発現率と重症度 【評価期間:最長78ヵ月】 |
| 副次的な評価項目 | 安全性/safety |
| 副次的な評価方法 | 死亡率 【評価期間:最長78ヵ月】 |
| 副次的な評価項目 | 安全性/safety |
| 副次的な評価方法 | 臨床検査値異常の発現率 【評価期間:最長78ヵ月】 |
| 予定試験期間 | 2019年12月4日~2026年6月30日 |
出典:臨床研究等提出・公開システムより