検査・診断
多発性骨髄腫の類縁疾患、検査、診断、ステージ分類、治療選択などをご紹介します。
多発性骨髄腫の検査
多発性骨髄腫の検査には、診断と治療方針を決定する目的で行われる治療前の検査と、治療の効果判定を目的として行われる検査があります。
診断と治療方針を決定する目的で行われる治療前の検査
多発性骨髄腫の疑いとなったときには、まず血液検査と画像検査が行われます。血液検査では、貧血、高カルシウム血症、腎機能の低下、総タンパク量の上昇、アルブミン値の低下などを調べます。画像検査では、全身X線検査、CT、MRI、PET-CTなどで、骨折や骨病変を調べます。
血液検査と画像検査で多発性骨髄腫の可能性が疑われるときは、確定診断のために骨髄検査が行われます。採取した骨髄中の形質細胞の比率が10%以上に増えていた場合、多発性骨髄腫と診断されます。また、血清中の免疫グロブリンを調べる血清免疫固定法検査※1や血清免疫グロブリン遊離軽鎖(FLC)検査※2も確定診断のために行われます。
※1血清免疫固定法検査:電気泳動法と呼ばれる分析法に抗原抗体反応を組み合わせた検査法で、血清中の微量なタンパク質(Mタンパクなど)を検出するときなどに用いられます。
※2血清免疫グロブリン遊離軽鎖(FLC)検査:正常な免疫グロブリンは2つの重鎖と2つの軽鎖からできています。多発性骨髄腫の異常な細胞から、軽鎖が過剰に産生されると、遊離した軽鎖が血液中にみられるようになります。この、遊離した軽鎖を調べる検査が、血清免疫グロブリン遊離軽鎖(FLC)検査法です。
治療前検査一覧
| 検査 | 内容 |
| 一般検査 | 検尿、便ヘモグロビン、血算・血液像、凝固検査、生化学・免疫検査(総タンパク、アルブミン、総ビリルビン、AST、ALT、ALP、LDH、アミラーゼ、アンモニア、尿酸、血糖、BUN、クレアチニン、ナトリウム、カリウム、カルシウム、タンパク分画、β2ミクログロブリン、CRP)、HBs抗原、HBc抗体、HBs抗体、HCV抗体、HIV抗体、胸部X線検査、心電図、動脈血酸素飽和度 |
| Mタンパクの同定と定量 | タンパク分画(血清、尿)、24時間尿タンパク定量 免疫電気泳動法(血清、尿)、または免疫固定法免疫グロブリン定量(IgG、IgA、IgD、IgM、IgE) 血清遊離軽鎖定量およびκ/λ比 |
| 骨髄形質細胞の増加 形質細胞腫の証明 | 骨髄穿刺・骨髄生検 フローサイトメトリーによる表面形質解析 染色体分析、FISH法 |
| 臓器障害の診断 | 全身骨X線検査(頭蓋骨:正・側、頸椎、胸椎、腰椎:正・側、肋骨:正面、骨盤骨:正面、左右の上腕骨:正面、左右の前腕骨:正面、左右の大腿骨:正面、左右の下腿骨:正面) 単純CT(頸部、胸部、腹部、骨盤部)脊椎、腸骨 MRI FDG-PET 骨代謝マーカー(尿中デオキシピリジノリン、血清NTx、尿CTx、骨型アルカリホスファターゼ、オステオカルシン)(保険適用に注意) クレアチニンクリアランス 心臓超音波検査 |
| その他の検査 (必要に応じ追加) | 生検(皮下組織、骨髄、口唇、胃、あるいは腎)、血液・血漿・血清粘稠度、眼底検査、チミジンキナーゼ、クリオグロブリン |
出典:一般社団法人日本血液学会編. 造血器腫瘍診療ガイドライン 2023年版.金原出版 第III章 骨髄腫、I 多発性骨髄腫 表2より作成
治療の効果判定を目的として行われる検査
治療効果判定では、Mタンパク(骨髄腫細胞から産生される免疫グロブリン)を測定するための血液検査や尿検査が行われます。全身の骨画像検査は、1年に1回受けることが推奨されています。また骨髄検査は、完全奏効の判断と非分泌型骨髄腫患者さんの効果判定のために行われます。これらの治療効果判定には、国際骨髄腫作業部会(IMWG)による統一効果判定基準が用いられています。
治療効果判定のために必要な検査
| 検査 | 内容 |
| Mタンパク量測定のための検査 | 血清Mタンパク量は、血清タンパク電気泳動を行い濃度測定で定量 ただしIgA型のようにMタンパクがβ分画にあるような場合は、血清タンパク電気泳動の信頼性が低いため、IgAの絶対値をMタンパク量として用いる 尿中Mタンパク量は、24時間尿の尿タンパク電気泳動法で測定 随時尿や24時間尿を用いたκ、λ軽鎖の定量検査は信頼に値せず、推奨できない |
| 測定可能病変の定義 | すべてのカテゴリおよび、完全奏効を除くサブカテゴリーの効果判定には、下記の測定可能病変のうち、最低1つが必要 血清Mタンパク≧1g/dL 尿中Mタンパク≧200mg/24時間 血清遊離軽鎖のκ/λ比が異常、かつMタンパクに一致する FLC値≧10mg/dL 完全奏効の効果判定を行う際は、上記の3つの測定可能病変のどれかが必要、厳格な完全奏効だけは上記3つのどれもない場合であっても判定可能 |
| 部分奏効もしくは安定規準の判断のためのフォローアップ | 新規治療開始後1年間は、月1回のフォローアップ 1年経過後は2か月ごとのフォローアップが推奨 測定可能病変がある場合は、血清タンパク電気泳動とタンパク白電気泳動で両者をフォローアップすることが必要 完全奏効判定を除き、血清タンパク電気泳動のみでしか測定可能病変がない場合は、血清タンパク電気泳動のみでのフォローアップ 同様にタンパク白電気泳動のみでしか測定可能病変がない場合は、タンパク白電気泳動のみでのフォローアップ 血清タンパク電動泳動もしくはタンパク白電気泳動、あるいはその両者の測定可能病変がある場合は、これら2種類のMタンパク測定に基づいて効果判定を行うべきであって、遊離L鎖測定に基づく効果判定は行わない 遊離L鎖を用いた効果判定は、あくまで血清タンパク電気泳動やタンパク白電気泳動においてMタンパク量測定が行えないときに用いる場合と、厳格な完全奏効のカテゴリを満たすか否かの判断に対して用いられるもの 完全奏効の判定には、必ず血清と尿の両者の免疫固定法が行われ、治療前のMタンパク量にかかわらず両者とも陰性であることを確認 治療前にタンパク白電気泳動が陰性であった場合でも完全奏効の確認のためには再度タンパク白電気泳動検査を行う 全身骨の画像検査は、臨床症状がない限りは効果判定目的に行う必要はない 一般臨床では、年に1回は実施することを推奨 骨髄検査は、完全奏効カテゴリの判断と非分泌型骨髄腫の効果判定に限って必要 |
出典:一般社団法人日本血液学会編. 造血器腫瘍診療ガイドライン 2023年版.金原出版 第III章 骨髄腫、I 多発性骨髄腫 表5より作成
多発性骨髄腫の類縁疾患と診断
多発性骨髄腫を含む形質細胞腫瘍の診断では、すぐに治療を必要としない安定した患者さんと、すぐに治療が必要な患者さんの見極めが重要とされます。高カルシウム血症、腎不全、貧血、骨病変のうち1つ以上の症状がある多発性骨髄腫の患者さんは、すぐに治療が必要で、全身化学療法の対象となります。また、適切な治療のために特に見極めが重要なのは「意義不明の単クローン性高ガンマグロブリン血症(MGUS)」、症状のない「くすぶり型多発性骨髄腫」、症状のある「多発性骨髄腫」などの病型です。
くすぶり型多発性骨髄腫の中でも、検査の結果から「2年以内に80%以上の確率で、症状のある多発性骨髄腫に移行する可能性がある」となった患者さんは、症状のある(症候性の)多発性骨髄腫に分類されます。しかし、一定割合以上の形質細胞が検出されても症状が現れないこともあり、直ちに治療となるか経過観察となるかは患者さんごとに判断されます。
以下の2つの基準を満たす場合、多発性骨髄腫(症候性)と診断されます。
(1)1つの細胞に由来する形質細胞が骨髄の中に10%以上ある(クローナルな骨髄中形質細胞≧10%)か、生検にて診断された骨性または軟部組織の形質細胞腫を認める
(2)骨髄腫診断事象の1つ以上、またはバイオマーカーの1つ以上を満たす
多発性骨髄腫の類縁疾患は、国際骨髄腫作業部会による診断基準により分類されます。
国際骨髄腫作業部会による診断基準
| 疾患名 | 基準 | 判定 |
|---|---|---|
| 非IgM型意義不明の単クローン性ガンマグロブリン血症 | (1)血清中非IgM型Mタンパク<3g/dL (2)クローナルな骨髄中形質細胞<10% (3)臓器障害を認めない | (1)~(3)のすべてを満たす |
| IgM型意義不明の単クローン性ガンマグロブリン血症 | (1)血清中IgM型Mタンパク<3g/dL (2)骨髄中リンパ形質細胞浸潤<10% (3)次の症候を欠如(貧血、全身症状、過粘稠、リンパ節腫大、肝脾腫とそれ以外の臓器障害) | (1)~(3)のすべてを満たす |
| 軽鎖型意義不明の単クローン性ガンマグロブリン血症 | (1)血清遊離軽鎖比の異常(<0.26または>1.65) (2)該当する血清遊離軽鎖の増加 (3)免疫固定法にて重鎖発現を認めない (4)臓器障害(CRABまたはアミロイドーシス)を認めない (5)クローナルな骨髄中形質細胞<10% (6)尿中Mタンパク量<500mg/24時間 | (1)~(6)のすべてを満たす |
| 孤立性形質細胞腫(骨の/軟部組織の) | (1)生検にてクローナルな形質細胞から成る骨あるいは軟部組織の形質細胞腫の存在 (2)骨髄中にクローナルな形質細胞を認めない (3)孤立性形質細胞腫病変以外には骨X線、椎体および骨盤MRI(またはCT)で異常を認めない (4)臓器障害(CRAB)を認めない | (1)~(4)のすべてを満たす |
| 微小骨髄浸潤を有する孤立性形質細胞腫(骨の/軟部組織の) | (1)生検にてクローナルな形質細胞から成る骨あるいは軟部組織の形質細胞腫の存在 (2)骨髄中のクローナルな形質細胞<10% (3)孤立性形質細胞腫病変以外には骨X線、椎体および骨盤MRI(またはCT)で異常を認めない (4)臓器障害(CRAB)を認めない | (1)~(4)のすべてを満たす |
| くすぶり型(無症候性)多発性骨髄腫 | (1)血清中Mタンパク(IgGまたはIgA型)≧3g/dLまたは尿中Mタンパク≧500mg/24時間 (2)クローナルな骨髄中形質細胞が10%以上で60%未満 (3)骨髄腫診断事象またはアミロイドーシスを認めない | (1)または(2)に加えて(3)を満たす |
| (症候性)多発性骨髄腫(分泌型/非分泌型) | (1)クローナルな骨髄中形質細胞≧10%または生検にて診断された骨性または軟部組織の形質細胞腫を認める (2)骨髄腫診断事象の1つ以上、またはバイオマーカーの1つ以上を満たす | (1)と(2)の両者を満たす (1)の骨髄中形質細胞が10%未満の場合は、2か所以上の骨病変を認めることが必要 |
| 多発性形質細胞腫 | (1)血清または尿中にMタンパクを検出しないか、検出しても微量である (2)クローナルな形質細胞による2か所以上の形質細胞腫または骨破壊を認める (3)正常骨髄 (4)形質細胞腫病変以外の骨所見に異常を認めない (5)臓器障害(CRAB)を認めない | (1)~(5)のすべてを満たす |
| 形質細胞白血病 | (1)末梢血中形質細胞>2,000/μL (2)白血球分画中形質細胞比率≧20% | (1)と(2)を満たす |
出典:一般社団法人日本血液学会編. 造血器腫瘍診療ガイドライン 2023年版.金原出版 第III章 骨髄腫、I 多発性骨髄腫 表1より作成
多発性骨髄腫のリスクとステージ分類
多発性骨髄腫は、年齢、病型、ステージ、合併症、染色体異常などがリスク因子として挙げられます。染色体異常については、「17番欠失」「4:14番転座」「14:16番転座」があると予後が良くないと知られています。
ステージは1~3に分類され、ステージの判定基準として、血清β2ミクログロブリン値とアルブミン値のみによる国際病期分類(ISS)を用いることが推奨されています。さらに、治療にプロテアソーム阻害薬や免疫調整薬が使用可能となったことで予後が改善されたため、改訂国際病期分類(R-ISS)が作成されました。R-ISSでは、高リスクの染色体異常の有無や増殖能を反映する血清LDH濃度が追加されました。
ISSの判定基準
| ステージ | 判定基準 | 50%生存期間 |
|---|---|---|
| 1 | 血清β2ミクログロブリン<3.5mg/L 血清アルブミン≧3.5g/dL | 62か月 |
| 2 | 血清β2ミクログロブリン<3.5mg/Lで血清アルブミン<3.5g/dL 血清アルブミン値にかかわらず血清β2ミクログロブリン≧3.5mg/Lかつ<5.5mg/L | 44か月 |
| 3 | 血清β2ミクログロブリン≧5.5mg/L | 29か月 |
出典:一般社団法人日本血液学会編. 造血器腫瘍診療ガイドライン 2023年版.金原出版 第III章 骨髄腫、I 多発性骨髄腫、表3より作成
R-ISSの判定基準
| ステージ | 判定基準 | >
| 1 | ISSステージ1かつ「17番欠失」「4:14番転座」「14:16番転座」の染色体異常がなく、かつ血清LDH正常範囲 |
| 2 | ステージ1でもなく2でもない |
| 3 | ISSステージ3、かつ「17番欠失」「4:14番転座」「14:16番転座」の染色体異常あり、または血清LDH高値 |
出典:一般社団法人日本血液学会編. 造血器腫瘍診療ガイドライン 2023年版.金原出版 第III章 骨髄腫、I 多発性骨髄腫、表4より作成
多発性骨髄腫の治療選択
多発性骨髄腫は、骨髄腫細胞を破壊して病気の進行を遅らせ、できるだけ長く良好な生活の質を保つことを目標に、治療法選択が行われます。
症候性骨髄腫の前がん病態である「意義不明の単クローン性免疫グロブリン血症」や「くすぶり型多発性骨髄腫」に対しては、無治療経過観察が原則となり、症候性の多発性骨髄腫に進行した場合に治療が開始されます。
初回治療は、自家造血幹細胞移植の適応となるかどうかで、治療選択が異なります。自家造血幹細胞移植の適応条件は、65歳未満で重篤な合併症がなく、心肺機能が正常な患者さんです。65歳以上や重要臓器に障害がある患者さん、また、移植を拒否する患者さんには適応されません。65歳という年齢は目安で、患者さんごとの状態により決定されます。
未治療で自家造血幹細胞移植が適応となる患者さんの治療選択
自家造血幹細胞移植が適応となる患者さんでは、迅速で深い奏効が期待できる寛解導入療法としてボルテゾミブ(製品名:ベルケイド)を含む併用療法やレナリドミド(製品名:レブラミド)を含む併用療法が選択されます。BLd療法(ボルテゾミブ+レナリドミド+低用量デキサメタゾン)が推奨されていますが、患者さんの病態に応じて、Bd療法(ボルテゾミブ+低用量デキサメタゾン)、BCd療法(ボルテゾミブ+シクロホスファミド+低用量デキサメタゾン)、BTd療法(ボルテゾミブ+サリドマイド+低用量デキサメタゾン)、BAD療法(ボルテゾミブ+ドキソルビシン+デキサメタゾン)、Ld療法(レナリドミド+低用量デキサメタゾン)も推奨されています。
寛解導入療法後、自家造血幹細胞移植を行うために血液中の幹細胞が採取した後、大量メルファラン療法により導入療法で残ったがん細胞を死滅させてから、採取しておいた造血幹細胞の移植が行われます。高リスク染色体異常がある場合は、移植を2回行うタンデム自家造血幹細胞移植も治療選択肢の1つとなります。
自家造血幹細胞移植後の維持療法として、レナリドミドが推奨されています。2年間の継続投与が推奨されていますが、有害事象や二次がんの発生に留意しながら、病勢進行まで継続することが考慮されています。また、ステージ3の患者さんやレナリドミド維持療法が困難な患者さんに対しては、イキサゾミブによる維持療法が選択肢となります。
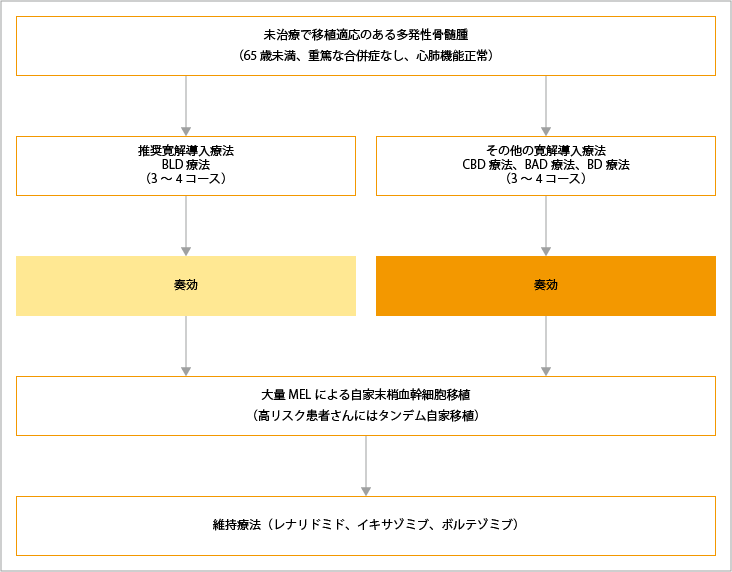
BLD療法:ボルテゾミブ+レナリドミド+デキサメタゾン
CBD療法:シクロホスファミド+ボルテゾミブ+デキサメタゾン
BAD療法:ボルテゾミブ+ドキソルビシン+デキサメタゾン
BD療法:ボルテゾミブ+デキサメタゾン
出典:一般社団法人日本血液学会編. 造血器腫瘍診療ガイドライン 2023年版.金原出版 第III章 骨髄腫、I 多発性骨髄腫、アルゴリズムより作成
自家造血幹細胞移植が適応とならない患者さんの治療選択
自家造血幹細胞移植が適応とならない患者さんに対しては、D-MPB療法(ダラツムマブ+メルファラン+プレドニゾロン+ボルテゾミブ)もしくは、DLd療法(ダラツムマブ+レナリドミド+低用量デキサメタゾン)が推奨されています。患者さんの病態に応じて、Bd療法(ボルテゾミブ+低用量デキサメタゾン)、MPB療法(メルファラン+プレドニゾロン+ボルテゾミブ)、Ld療法(レナリドミド+低用量デキサメタゾン)、減量BLd療法(ボルテゾミブ+レナリドミド+低用量デキサメタゾン)も治療選択の1つとなります。
D-MPB療法後に奏効が認められた場合は、10コース目からダラツムマブ単剤による維持療法が行われます。DLd療法後に奏効が認められた場合は、病勢進行もしくは許容できない有害事象が起こるまで継続治療が行われます。
ダラツムマブを含まないその他の治療後は、Ld療法およびレナリドミド維持療法が行われ、治療の継続期間は患者さんの病態に応じて決定されます。
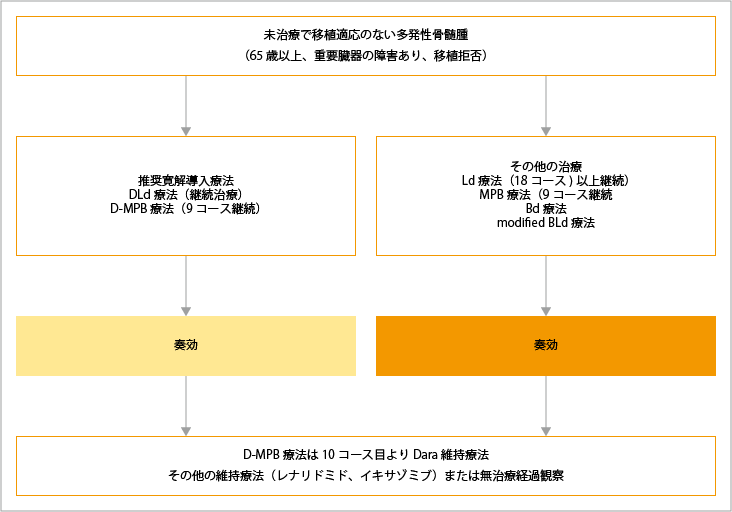
DLd療法:ダラツムマブ+レナリドミド+低用量デキサメタゾン
D-MPB療法;ダラツムマブ+メルファラン+プレドニゾロン+ボルテゾミブ
Ld療法:レナリドミド+低用量デキサメタゾン
MPB療法;メルファラン+プレドニゾロン+ボルテゾミブ
Bd療法:ボルテゾミブ+低用量デキサメタゾン
modified-BLd療法:減量(ボルテゾミブ+レナリドミド+低用量デキサメタゾン)
出典:一般社団法人日本血液学会編. 造血器腫瘍診療ガイドライン 2023年版.金原出版 第III章 骨髄腫、I 多発性骨髄腫 アルゴリズムより作成
参考文献:一般社団法人日本血液学会編. 造血器腫瘍診療ガイドライン 2023年版.金原出版